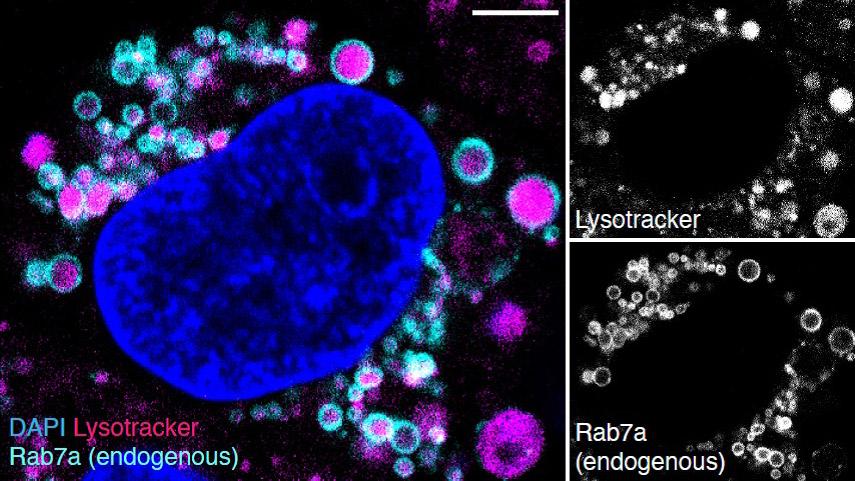
In vitro characterization of complex medicines
In recent years, the focus of medical discovery has shifted from traditional small molecule approaches to complex therapies such as nanomedicines and biologics. Prior to application in vivo, the molecular dynamics and mechanism of these novel therapies must be thoroughly characterized in vitro. This not only helps to ensure efficacy and safety, but also advances the predictive capability of in vitro models and provides an avenue for the rational design of new medicines. In this study, Medicines Discovery Catapult (MDC), a non-profit medicines discovery organization in the United Kingdom, and ZEISS have developed an approach that combines genome engineering, advanced microscopy, and computational image analysis to quantify the plasma membrane binding, internalization and trafficking of Herceptin, an antibody-based complex medicine. We note that this approach is broadly applicable to other classes of complex medicines, such as silica nanoparticles, lipid nanoparticles, nucleic acids, and antibody-drug conjugates.
Antibody-based therapeutics have made valuable contributions to the treatment of cancer, autoimmune conditions, and aging-related disorders. Key to antibody-based approaches is the existence of a plasma membrane-bound or extracellular protein target whose function is critical to disease etiology.
Antibodies exert several effects when targeted to plasma membrane-bound receptors. In addition to directly blocking the receptor’s natural ligand binding and preventing activation of the receptor’s signaling cascade, receptor-bound antibodies can also provide an epitope for subsequent binding of immune cells and stimulate the Antibody-Dependent Cell-mediated Cytotoxicity (ADCC), whereby the target cell (e.g., a cancer cell) is destroyed. Antibody-bound receptors can also be internalized from the cell’s surface by receptor-mediated endocytosis and then trafficked into the endolysosomal network. The latter process is key for a group of complex medicines called antibody-drug conjugates (ADC), which exploit the specificity of antibody-epitope binding to deliver a therapeutic payload intracellularly.